Integration with transcriptomics#
In this tutorial, we will continue working with the artery datasets from the previous section to demonstrate multi-modal analysis capabilities.
The GTEx dataset provides a unique opportunity for integrative analysis as it contains paired RNA-seq and histology data for the same tissue samples.
We begin by downloading the RNA-seq data (normalized TPM matrix) and the morphological features we computed in the previous sections.
from huggingface_hub import hf_hub_download
rna = hf_hub_download(
"rendeirolab/lazyslide-data", "GTEx_artery_RNA.h5ad", repo_type="dataset"
)
wsi_features = hf_hub_download(
"rendeirolab/lazyslide-data", "agg_conch_features.h5ad", repo_type="dataset"
)
import anndata as ad
import scanpy as sc
rna = ad.read_h5ad(rna)
wsi_features = ad.read_h5ad(wsi_features)
Calcification scoring#
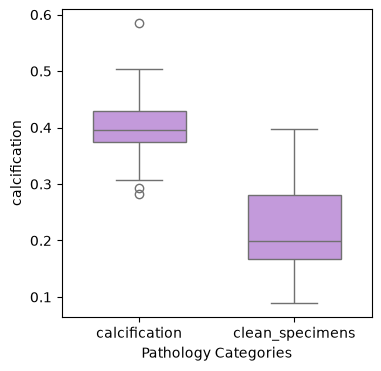
We first examine the distribution of calcification scores across different pathological categories to understand the morphological patterns in our dataset.
import seaborn as sns
sns.boxplot(
data=wsi_features.obs,
x="Pathology Categories",
y="calcification",
color="#C68FE6",
width=0.6,
)
<Axes: xlabel='Pathology Categories', ylabel='calcification'>
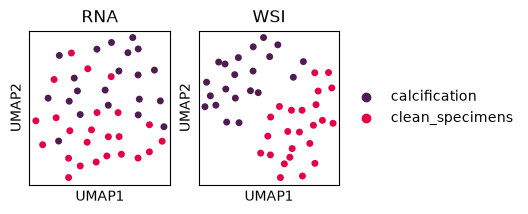
Visualization of RNA and Histological morphological features#
To compare the structure of our two data modalities, we will generate UMAP embeddings for both the RNA-seq data and the WSI morphological features. This visualization will help us understand how samples cluster in each modality and identify potential relationships between molecular and morphological patterns.
def run_umap(adata):
sc.pp.scale(adata)
sc.pp.pca(adata)
sc.pp.neighbors(adata)
sc.tl.umap(adata)
run_umap(rna)
run_umap(wsi_features)
import matplotlib.pyplot as plt
fig, axes = plt.subplots(ncols=2, figsize=(4, 2))
opts = dict(
color="Pathology Categories", palette=["#4F1C51", "#E50046"], size=100, show=False
)
sc.pl.umap(rna, ax=axes[0], legend_loc=None, **opts)
sc.pl.umap(wsi_features, ax=axes[1], **opts)
axes[0].set_title("RNA")
axes[1].set_title("WSI")
plt.show(fig)
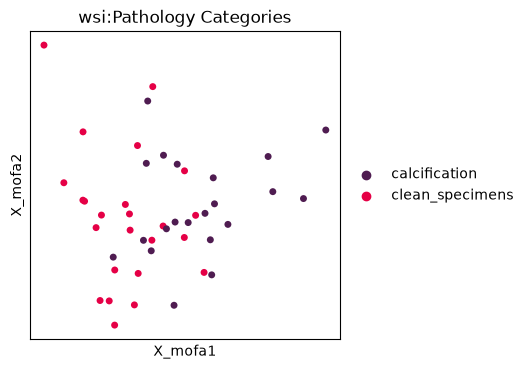
Understanding variance across multiple modalities#
Multi-Omics Factor Analysis (MOFA) is a well-established method for identifying latent factors that explain variance across multiple data modalities. This approach allows us to decompose the variance in our RNA-seq and WSI feature data into shared and modality-specific components.
Prerequisites: You will need to install muon and mofapy2 to execute the MOFA analysis.
import muon as mu
mdata = mu.MuData({"wsi": wsi_features, "rna": rna})
mu.tl.mofa(mdata, verbose=False)
#########################################################
### __ __ ____ ______ ###
### | \/ |/ __ \| ____/\ _ ###
### | \ / | | | | |__ / \ _| |_ ###
### | |\/| | | | | __/ /\ \_ _| ###
### | | | | |__| | | / ____ \|_| ###
### |_| |_|\____/|_|/_/ \_\ ###
### ###
#########################################################
Loaded view='wsi' group='group1' with N=45 samples and D=512 features...
Loaded view='rna' group='group1' with N=45 samples and D=31439 features...
Model options:
- Automatic Relevance Determination prior on the factors: True
- Automatic Relevance Determination prior on the weights: True
- Spike-and-slab prior on the factors: False
- Spike-and-slab prior on the weights: True
Likelihoods:
- View 0 (wsi): gaussian
- View 1 (rna): gaussian
######################################
## Training the model with seed 1 ##
######################################
Converged!
#######################
## Training finished ##
#######################
Saving model in /tmp/mofa_20260619-213011.hdf5...
Saved MOFA embeddings in .obsm['X_mofa'] slot and their loadings in .varm['LFs'].
mu.pl.mofa(
mdata, color="wsi:Pathology Categories", palette=["#4F1C51", "#E50046"], size=100
)
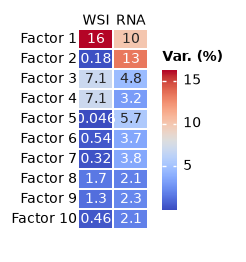
import numpy as np
import marsilea as ma
loadings = mdata.uns["mofa"]["variance"]
h = ma.Heatmap(
np.array([loadings["wsi"], loadings["rna"]]).T,
annot=True,
cbar_kws={"width": 1, "height": 10},
width=0.7,
height=2,
linewidth=0.1,
label="Var. (%)",
)
h.add_top(ma.plotter.Labels(["WSI", "RNA"], rotation=0))
h.add_left(ma.plotter.Labels([f"Factor {i}" for i in np.arange(10) + 1]))
h.add_legends(box_padding=1)
h.render()
<marsilea.heatmap.Heatmap at 0x7f5fd8f85040>
Linking modalities through gene-morphology associations#
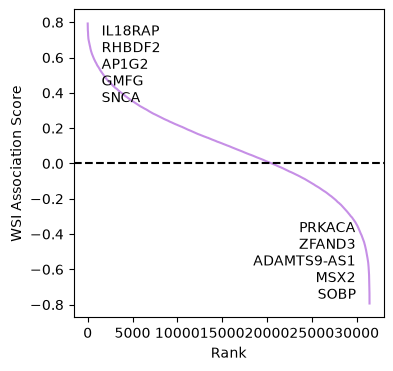
To integrate WSI morphological features with bulk RNA-seq data, we employ the RNALinker tool to identify genes whose expression levels correlate with specific morphological features. In this case, we focus on genes associated with the calcification score, using Spearman correlation to capture monotonic relationships between gene expression and morphological patterns.
import lazyslide as zs
linker = zs.tl.RNALinker(wsi_features, rna)
linker.associate(method="spearman", score_key="calcification")
linker.plot_rank(gene_name="Description")
genes = linker.associated_genes(100, gene_name="Description")
Functional enrichment analysis#
To interpret the biological significance of genes associated with calcification, we perform functional enrichment analysis using decoupler. We prepare the input data by organizing the top and bottom-ranked genes (based on their association with calcification) into a matrix format suitable for pathway analysis.
import pandas as pd
decoupler_input = pd.concat([genes["top"], genes["bottom"]]).T
decoupler_input
| Description | IL18RAP | RHBDF2 | AP1G2 | GMFG | SNCA | PLA2G7 | ENSG00000234389 | NPL | ARL4C | RASAL3 | ... | ENSG00000268798 | RBPMS2 | KCTD1 | MCAM | MIR325HG | PRKACA | ZFAND3 | ADAMTS9-AS1 | MSX2 | SOBP |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| association_score | 0.793149 | 0.781159 | 0.781028 | 0.780896 | 0.778656 | 0.7764 | 0.77632 | 0.774967 | 0.769565 | 0.767062 | ... | -0.69776 | -0.699209 | -0.703821 | -0.731489 | -0.735968 | -0.744005 | -0.748617 | -0.753491 | -0.77668 | -0.793412 |
1 rows × 200 columns
import decoupler as dc
msigdb = dc.op.resource("MSigDB") # Use MSigDB
markers = msigdb.rename(columns={"geneset": "source", "genesymbol": "target"})[
msigdb["collection"] == "go_molecular_function"
][["source", "target"]].drop_duplicates() # Filter for GO Molecular Function
acts, padj = dc.mt.ulm(data=decoupler_input, net=markers, tmin=0)
dc.pl.barplot(data=acts, name="association_score", top=25, figsize=(3, 5))

We can run the same analysis on RNA alone to see the difference
Comparison: RNA-only analysis#
To demonstrate the added value of integrating morphological features, we perform the same enrichment analysis using only RNA-seq data.
import scanpy as sc
sc.tl.rank_genes_groups(rna, "Pathology Categories", method="wilcoxon")
df = sc.get.rank_genes_groups_df(rna, "calcification").sort_values("scores")
decoupler_input_rna = pd.concat([df.head(100), df.tail(100)])[["names", "scores"]]
decoupler_input_rna["names"] = decoupler_input_rna["names"].map(
rna.var["Description"].to_dict()
)
decoupler_input_rna = decoupler_input_rna.set_index("names").T
acts, padj = dc.mt.ulm(data=decoupler_input_rna, net=markers, tmin=0)
dc.pl.barplot(data=acts, name="scores", top=25, figsize=(3, 5))

Conclusions#
The integration of WSI morphological features with transcriptomic data yields enrichment results that are more specifically relevant to calcification processes. Notably, our analysis reveals functionally relevant changes such as the downregulation of calmodulin binding pathways.
In contrast, enrichment analysis performed solely on RNA-seq data fails to identify molecular activities that are specifically associated with calcification processes. This demonstrates the significant value of incorporating morphological features from histological images to enhance the biological interpretation of transcriptomic data.